My research focuses on generative modelling for structural biology and machine learning methods for bioinformatics. Current interests include score-based diffusion, flow-matching, and protein language model representations, with the broader goal of accelerating computational tools for protein engineering and drug discovery.
Publications
Below is a list of my publications in reversed chronological order.
2026
-
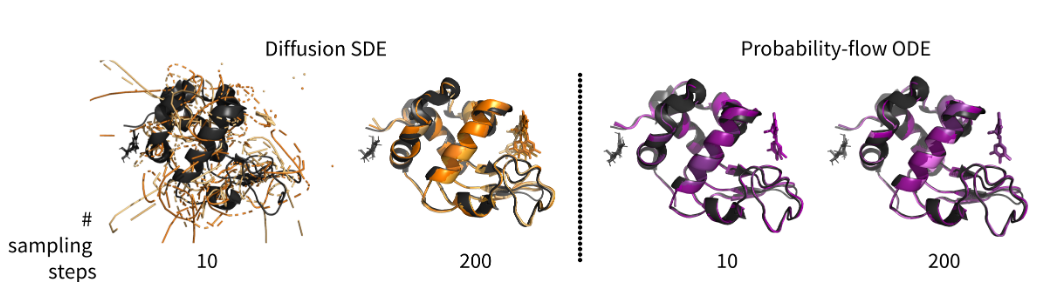
Converting diffusions to flows accelerates sampling and suggests over-conditioning of co-folding models on sequence
Nele P. Quast,
Niklas Abraham, Aaron Schöne, Fergus Imrie, Matthew I. J. Raybould, Yee Whye Teh, and Charlotte Deane
In ICLR 2026 Workshop on Learning Meaningful Representations of Life (LMRL) 2026
Deep generative models can predict protein structures from sequence with high accuracy; however, sampling from these models remains computationally burdensome, with current protocols using hundreds of iterations through the trained model to obtain a final predicted structure. To accelerate sampling and improve the interpretability of the prediction trajectories, we convert the stochastic diffusion sampling process into a deterministic flow process. We show that the conversion of pre-trained, diffusion-based structure prediction models to probability-flow ODEs yields equivalent performance on the FoldBench benchmark alongside a 20x sampling speed-up. Furthermore, we demonstrate the effects on prediction diversity and use the intermediate predictions made along the de-noising trajectory to show that deep generative structure prediction methods are strongly conditioned on the sequence and MSA embeddings, appearing to make predictions with weak sensitivity to the noise initialisation. Finally, we discuss the implications of strong sequence conditioning for generative protein structure prediction and protein design, as well as pointing to future experiments that build on our initial results.
-
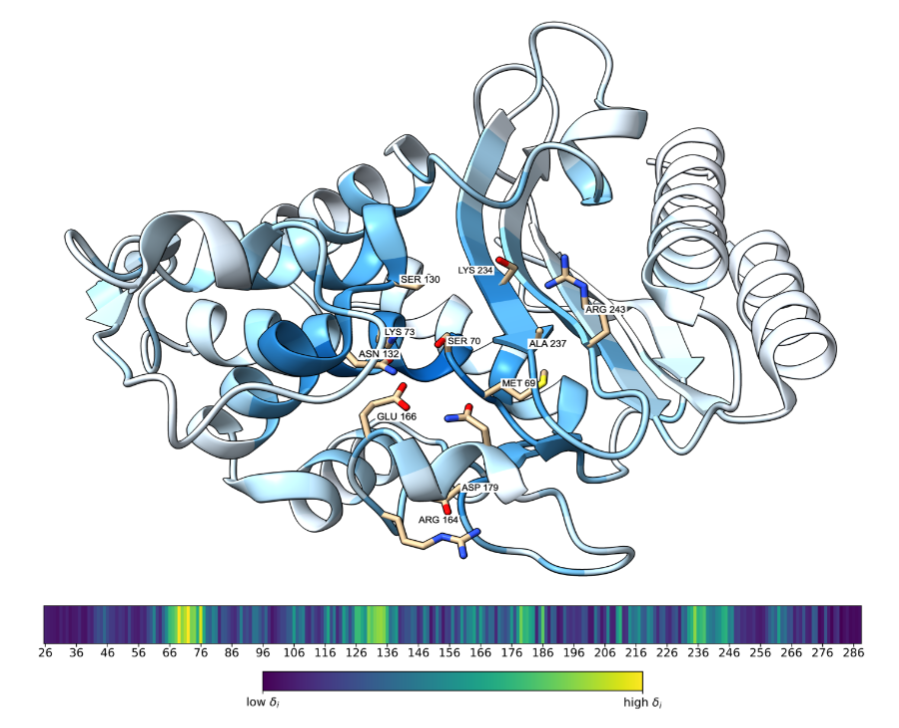
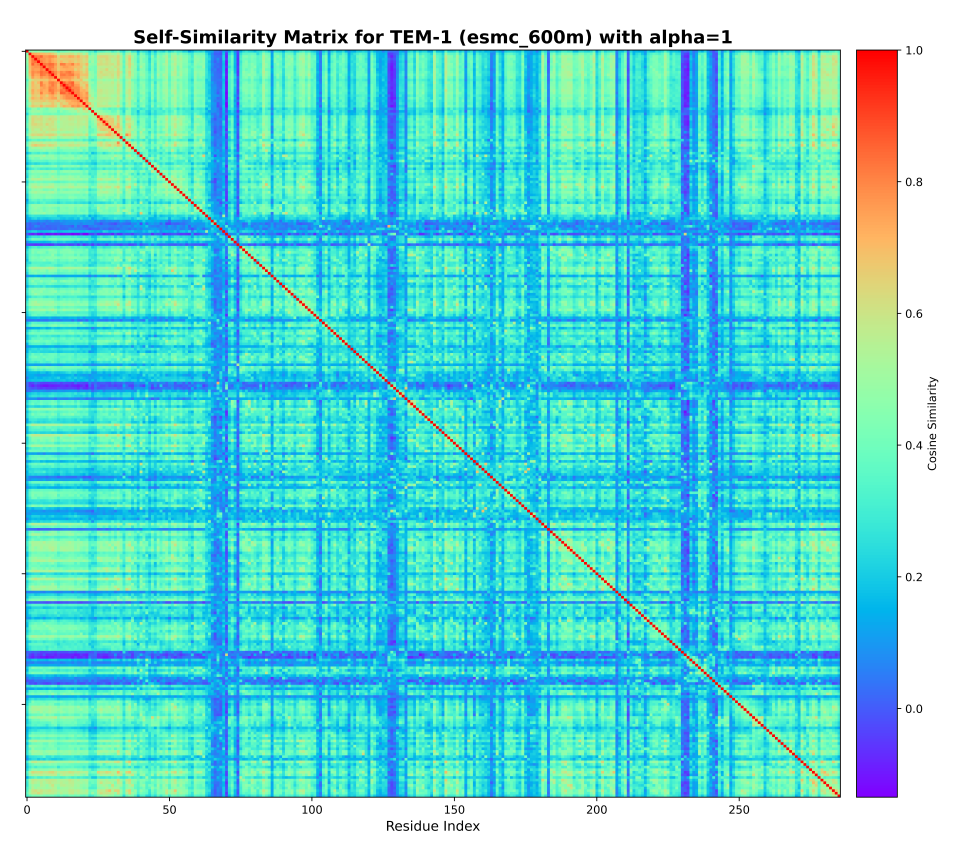
Geometric encoding of enzymatic mechanism in protein language model representations
Jan Range,
Niklas Abraham, and Jürgen Pleiss
Manuscript in preparation 2026
Understanding how protein language models encode biological function remains a fundamental challenge despite their remarkable empirical success. Sequence-level inference typically reduces per-residue embeddings to a single vector via mean pooling, disregarding the internal geometry of the token cloud. We propose that functionally critical residues are geometric outliers within this cloud: their embeddings deviate from the within-protein mean in direction rather than magnitude, reflecting the superposition of mechanism-specific features learned during masked-token pretraining against a structurally generic majority. Here we show that cosine dissimilarity from the within-protein mean identifies these residues without supervision, and that the subspace spanned by their embeddings carries precise functional information. Across the SwissProt proteome this subspace is enriched for active-site and binding annotations and localises to ligand-binding sites in experimentally resolved structures. Its principal direction encodes catalytic mechanism at sub-subclass resolution on sequence-redundancy-filtered enzymes and rotates in a fitness-predictable manner under single amino acid substitutions, without reliance on evolutionary alignments, structural data, or likelihood scoring. These results reveal that protein language models implicitly organise functional information as a geometric minority, separable from the structural majority by direction alone.
2025
-
Predicting β-lactamase resistance phenotypes from sequence using protein language models
Niklas Abraham
Bachelor’s Thesis, University of Stuttgart 2025
Protein language models (PLMs) have emerged as powerful tools for predicting protein structures from sequence. However, their ability to predict resistance phenotypes of β-lactamase enzymes remains unexplored. We propose a novel approach to predict β-lactamase resistance phenotypes from sequence using protein language models. We train a protein language model on a dataset of β-lactamase enzymes and their resistance phenotypes, and evaluate its performance on a dataset of β-lactamase enzymes and their resistance phenotypes. We find that the protein language model is able to predict β-lactamase resistance phenotypes with high accuracy, and that the model is able to predict resistance phenotypes that are not present in the training data. We also find that the model is able to predict resistance phenotypes that are present in the training data, but with lower accuracy. We conclude that protein language models are a powerful tool for predicting β-lactamase resistance phenotypes from sequence.